Publications
Explore our technology and the science behind it through these papers in highly-regarded publications.
Sygnomics in the news
Explore these articles to learn more about our trajectory as a company.
October 18th, 2023
The Andy Hill Cancer Research Endowment (CARE) Fund awarded a $2 million Life Science Startup Catalyst grant to SYGNOMICS, a personalized precision oncology startup that helps oncologists improve patient outcomes by accurately predicting each patient’s risk of disease progression and identifying specific therapies that could be effective against their cancer.
“This funding is vital to help us advance and deliver personalized precision oncology solutions. We are grateful to CARE for this award.”
—Nitin Baliga, Ph.D., the principal investigator of the CARE grant and co-founder and chief science officer of SYGNOMICS, a spin-out from the Institute for Systems Biology (ISB).
“This grant will help us accelerate our product development and clinical trial partnerships to support more oncologists and patients and address our large target market segment at the intersection of cancer genomic testing-based therapy selection ($1.4B), cancer biomarker market ($28.2B by 2026), and the real-world evidence market ($2B by 2027).”
—Ashutosh Tiwary, co-founder and CEO of SYGNOMICS.
Currently, oncologists rely on limited information from mutation-based genomic testing of a patient’s tumor biopsy to inform the severity of their patient’s disease and select treatment options for them. While these tests generate gene-expression data, unfortunately, current testing providers and oncologists do not fully utilize this information to improve the accuracy of predicting the risk of disease progression and selecting appropriate therapies matched to the characteristics of an individual patient’s cancer. Using patented systems-biology-based network analysis of the patient’s RNA sequence data, Sygnomics identifies specific disease pathways driving their cancer, which can then be used to (a) accurately predict the risk of disease progression, (b) recommend therapies that might help, and (c) identify therapies that are unlikely to help the patient. Delivered as a Sygnomics companion Laboratory Developed Test (LDT) report to existing genomic testing for each patient, this helps oncologists improve patient outcomes. Using real-world evidence (RWE) generated from oncologists treating patients, we will subsequently develop systems-biology-based biomarkers that can stratify patient populations with specific characteristics that may benefit from a specific therapy. These biomarkers can help pharmaceutical companies get existing drugs approved for new indications rapidly as well as improve outcome-based reimbursement for those drugs.
SYGNAL (SYstems Genetics Network AnaLysis) developed over a decade in Dr. Nitin Baliga’s lab at ISB and published in leading peer-reviewed journals, is the foundational technology for Sygnomics. Founded in December 2020 by Nitin and Ashutosh Tiwary, the Sygnomics team has a strong background in the science (computational biology), its clinical application (precision oncology), the delivery technology (software and cloud services), and serial entrepreneurship (in software and biotechnology). Nitin’s team (including Dr. Serdar Turkarslan) developed the original technology at the Institute for Systems Biology where he is a professor and SVP, and they lead the scientific development at Sygnomics. After spending 30+ years in the technology industry, where Ashutosh founded, built, and sold two technology startups and built significant businesses in the technology industry, he joined ISB as an entrepreneur-in-residence and helped shape the technology, its clinical application, and its business model. Dr. Anoop Patel is a neurosurgical oncologist at Duke University treating glioblastoma patients in his clinical practice. At Sygnomics, he is responsible for all aspects of translational medicine and its clinical applications.
Thanks to the Washington State legislature’s 2022 commitment to improve the lives of Washingtonians through an investment in cancer research, CARE awarded 32 grants totaling nearly $31 million. SYGNOMICS was one of 16 life sciences start-up and development awards supporting early-stage companies and innovative researchers to translate promising research into the development of tools, devices, or therapeutics related to cancer. CARE announced 16 additional awards in the areas of population health, inclusion and diversity in clinical trials, and shared resources and infrastructure.
January 17th, 2023
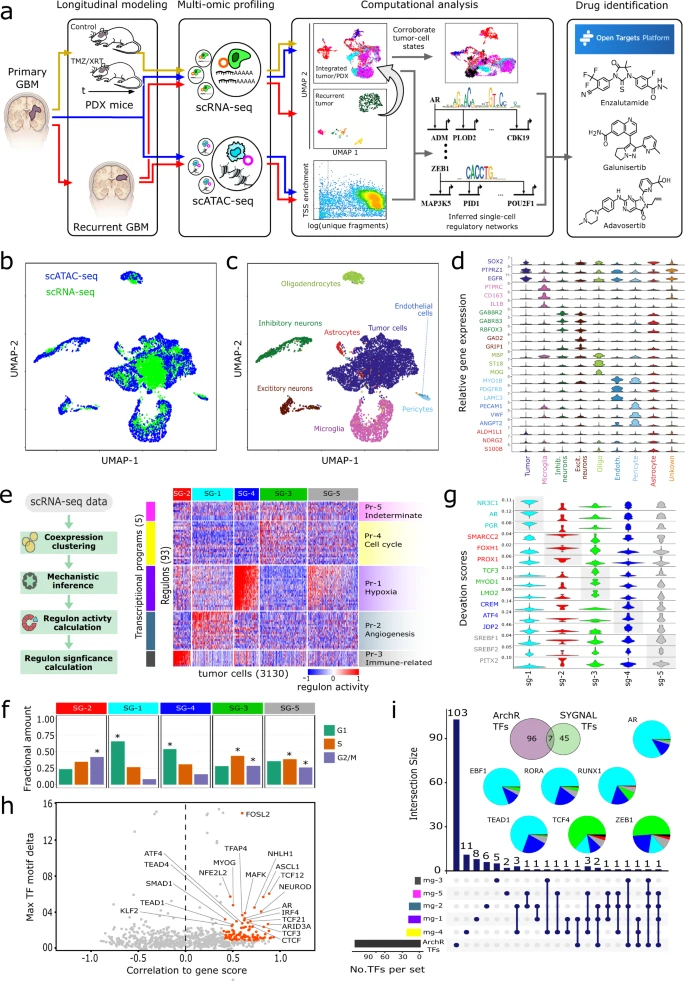
Glioblastoma (GBM) is a heterogeneous tumor made up of cell states that evolve over time. Here, we modeled tumor evolutionary trajectories during standard-of-care treatment using multi-omic single-cell analysis of a primary tumor sample, corresponding mouse xenografts subjected to standard of care therapy, and recurrent tumor at autopsy. We mined the multi-omic data with single-cell SYstems Genetics Network AnaLysis (scSYGNAL) to identify a network of 52 regulators that mediate treatment-induced shifts in xenograft tumor-cell states that were also reflected in recurrence. By integrating scSYGNAL-derived regulatory network information with transcription factor accessibility deviations derived from single-cell ATAC-seq data, we developed consensus networks that modulate cell state transitions across subpopulations of primary and recurrent tumor cells. Finally, by matching targeted therapies to active regulatory networks underlying tumor evolutionary trajectories, we provide a framework for applying single-cell-based precision medicine approaches to an individual patient in a concurrent, adjuvant, or recurrent setting.
A single-cell based precision medicine approach using glioblastoma patient-specific models
James H. Park, Abdullah H. Feroze, Samuel N. Emerson, Anca B. Mihalas, C. Dirk Keene, Patrick J. Cimino, Adrian Lopez Garcia de Lomana, Kavya Kannan, Wei-Ju Wu, Serdar Turkarslan, Nitin S. Baliga & Anoop P. Patel
npj Precision Oncology volume 6, Article number: 55 (2022)
November 20th, 2022
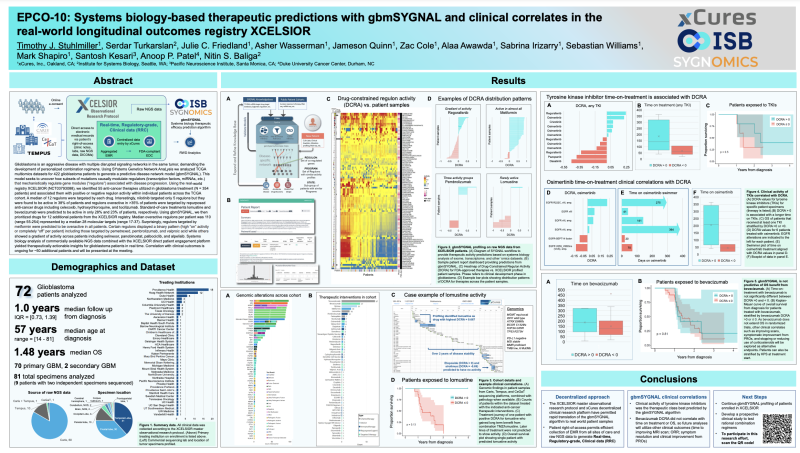
Sygnomics in collaboration with xCures presented a poster at 2021 SNO Annual Meeting to demonstrate how systems biology-based predictive cancer models for GBM in combination with real-world data registry can help decision-making and treatment options for CNS cancer patients.
Using SYstems Genetics Network AnaLysis team analyzed TCGA multiomics datasets for 422 glioblastoma patients to generate a predictive disease network model (gbmSYGNAL). This model seeks to uncover how subsets of mutations causally modulate regulators (transcription factors, miRNAs, etc.) that mechanistically regulate gene modules (“regulons”) associated with disease progression.
Using xCures’ real-world registry XCELSIOR (NCT03793088), team identified 55 anti-cancer therapies utilized in glioblastoma treatment (N = 354 patients) and associated them with positive or negative regulon activity within individual patients across the TCGA cohort. A median of 12 regulons were targeted by each drug. Interestingly, nilotinib targeted only 5 regulons but they were found to be active in 38% of patients and regulons overactive in >35% of patients were targeted by repurposed anti-cancer drugs including celecoxib, hydroxychloroquine, and tocilizumab. Standard-of-care treatments lomustine and bevacizumab were predicted to be active in only 28% and 23% of patients, respectively.
Using gbmSYGNAL, team then prioritized drugs for 12 additional patients from the XCELSIOR registry. Median overactive regulons per patient was 110 (range 55-254) represented by a median 28 molecular targets (range 17-37). Surprisingly, regulons targeted by metformin were predicted to be overactive in all patients. Certain regulons displayed a binary pattern (high “on” activity or completely “off” per patient) including those targeted by pemetrexed, pembrolizumab, and valproic acid while others showed a gradient of activity across patients including selinexor, panobinostat, palbociclib, and alpelisib. Systems biology analysis of commercially available NGS data combined with the XCELSIOR direct patient engagement platform yielded therapeutically actionable insights for glioblastoma patients in real time. Correlation with clinical outcomes is ongoing for ~50 additional patients and will be presented at the meeting.
EPCO-10: Systems biology-based therapeutic predictions with gbmSYGNAL and clinical correlates in the real-world longitudinal outcomes registry XCELSIOR
Timothy J. Stuhlmiller, Serdar Turkarslan, Julie C. Friedland, Asher Wasserman, Jameson Quinn, Zac Cole, Alaa Awawda, Sabrina Irizarry, Sebastian Williams, Mark Shapiro, Santosh Kesari, Anoop P. Patel, Nitin S. Baliga